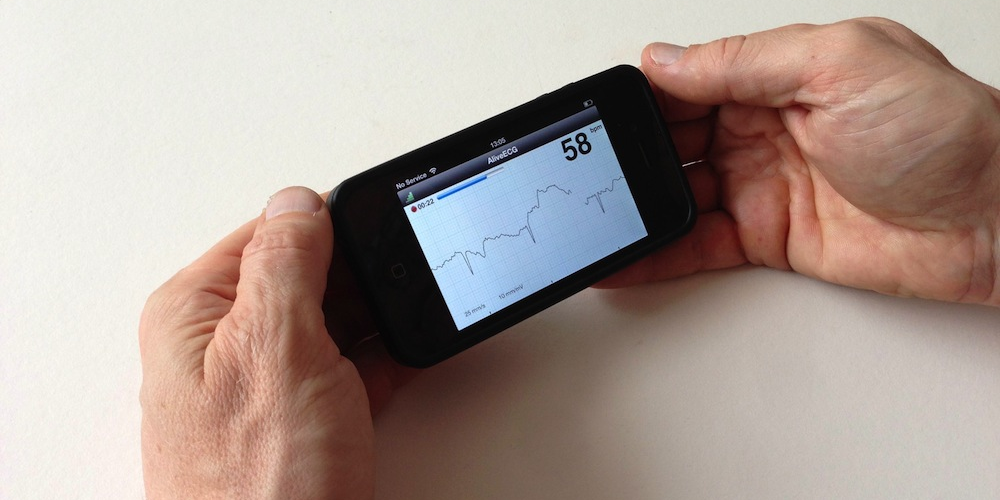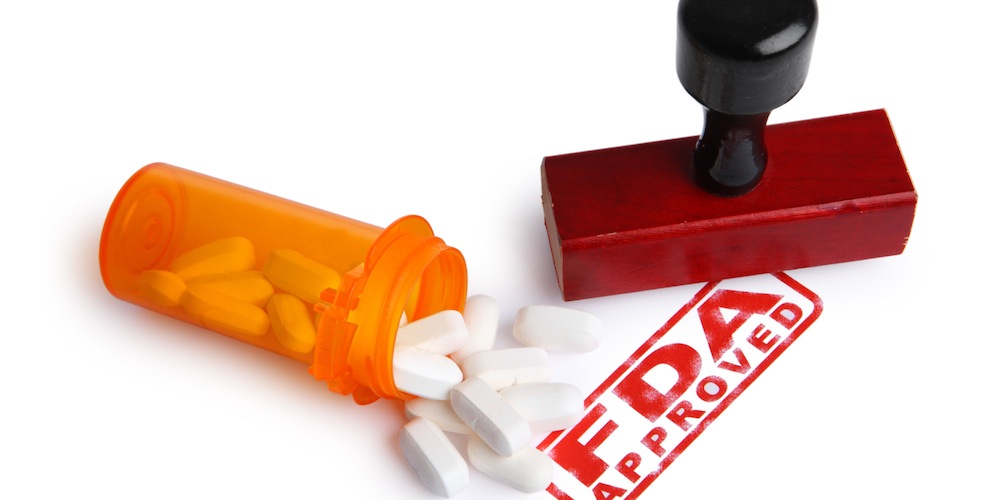
[칼럼] 올해 발표된 FDA의 디지털 헬스 규제 개선
*본 칼럼은 청년의사에 제가 기고한 글입니다. 원글은 여기에서 보실 수 있습니다. 지면에는 분량 때문에 요약된 글의 원래 버전을 올려드립니다. 지난 설 연휴 미국에서는 놀라운 소식이 들렸다. FDA가 23andMe의 개인 직접 판매 방식의 유전자 테스트를 최초로 승인한 것이다. 2013년 11월 정확성 검증 미비를 근거로 이 회사의 개인 유전정보 분석 서비스에 대한 판매 금지 명령을 내린지 14개월 만이다. 이번 조치로 개인 유전 정보 분석의 시장의 확대가 빨라질 것이라는 기대가 높다. FDA에 따르면 이번 심사 과정에서 이 기업의 유전자 테스트는 클래스 II 로 분류되었다. 위험도가 다소 낮은 의료기기에 부과되는 이 등급의 기기는 별도의 심사 과정 필요 없이, 사전 등록만 하면 시장에서 판매할 수 있다. 따라서 […]